

2020年11月:本课题组李超男博士开发的微生物组数据库:一站式环境基因组学数据云平台更新啦!
2021-08-20 14:46
微生物生态学学科的快速发展产生了大量的序列数据集,目前一般存储在NCBI、ENA和MG-RAST等国际生物信息学数据库。但是这些数据库缺少用于检索、整合和重新分析多个独立数据集所必需的信息,导致现有数据集利用率低、使用时间成本高等问题。此外,微生物组数据分析涉及大量的软件和多种编程语言,学习时间成本高。中国科学院成都生物研究所李香真团队经过近3年的开发与迭代,微生物组数据库V1.3版本现已正式上线(http://egcloud.cib.cn,推荐使用Chrome浏览器访问)。当前版本更新了生物信息分析流程、增加了生物信息模块分析、实时多元生态统计分析和可视化功能。
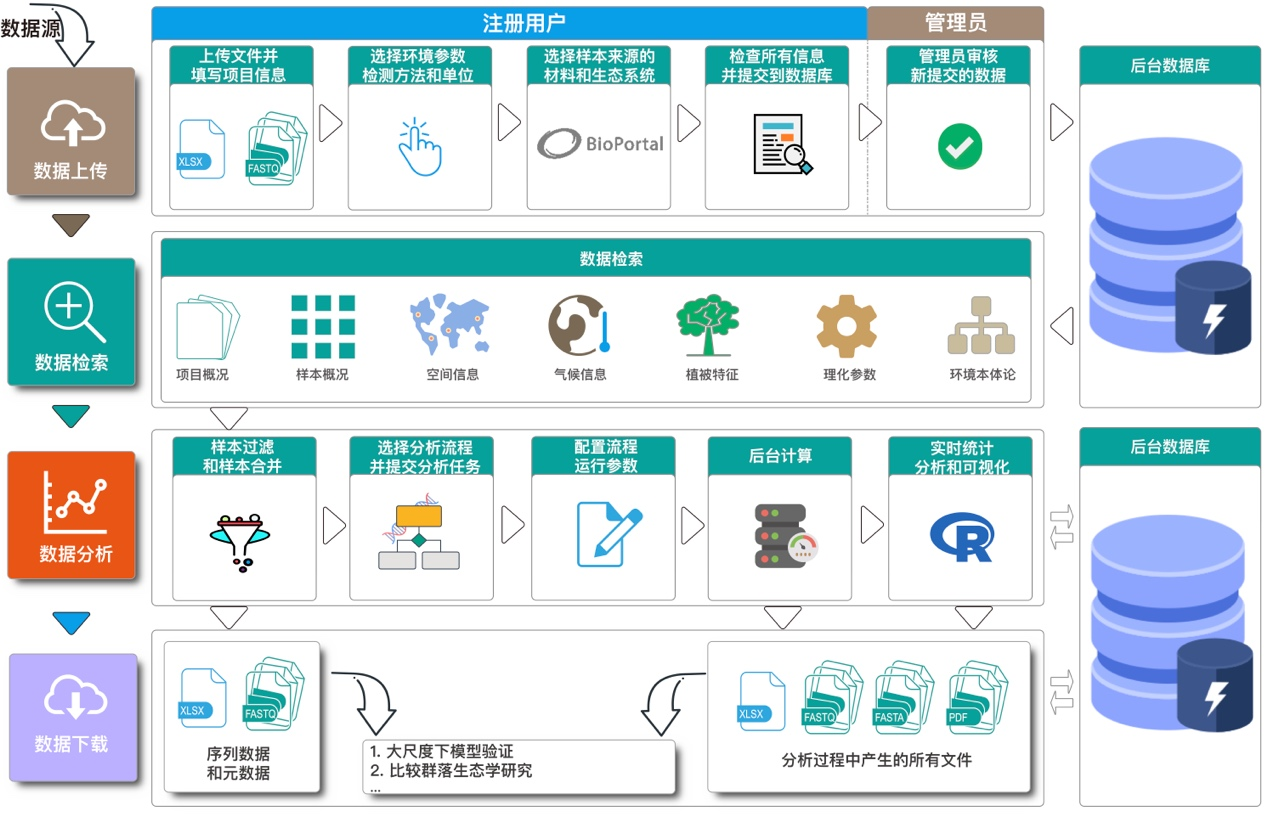
微生物组数据库的一大特点是同时收集微生物测序数据和与之关联的环境参数信息,为用户提供数据的存储、检索和分析服务。基于测序样本详细的环境参数信息,我们实现了对具有特定属性的样本的精确查询,可提高数据的使用率,降低数据收集的时间成本,为大尺度下微生物生态学研究提供了优质的数据源。微生物组数据库不仅是一个专业的实体数据库,也是一个一站式比较群落生态学研究的云平台。我们为所有注册用户免费提供“数据存储—检索—生物信息学分析—统计分析和可视化”一站式服务,让用户能够轻易地获取用于meta分析的数据集,不用写代码也能进行生物信息分析、统计分析和可视化。
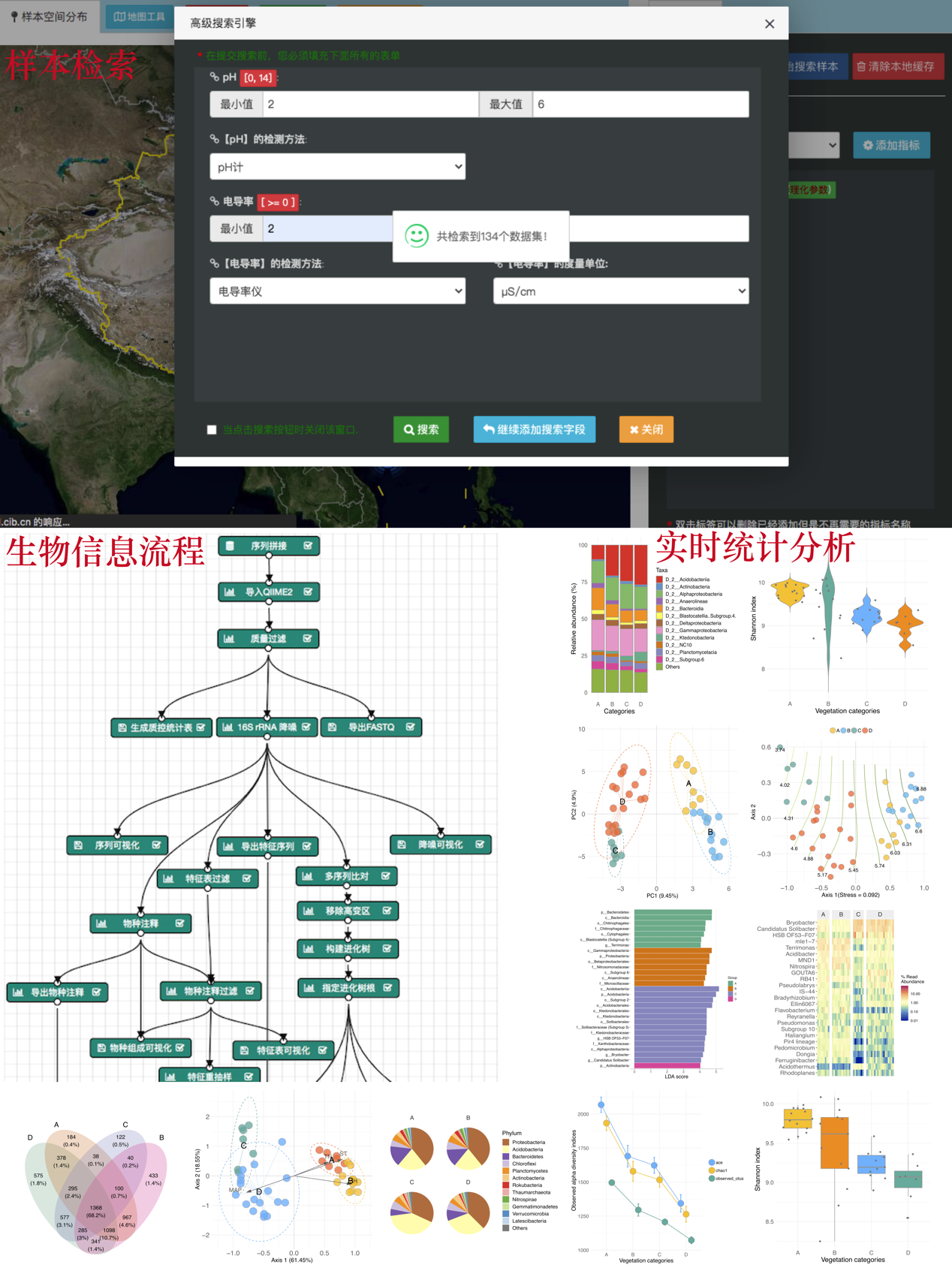
目前,微生物组数据库已经收录了包括16S rRNA基因、18S rRNA基因、ITS、功能基因(nifH、phoD、pmoA等)和宏基因组在内的高质量数据集3529套,其中包括序列文件、样本概况、空间信息、气候特征、植被信息和理化特征信息。数据库中收录的数据有最多两年的保护期(取决于提交数据的用户),到期后会自动释放,任何注册用户均可免费使用这些数据集进行科学研究。同时我们也希望科研界广大同僚们能够将已发表的数据集提交到微生物数据库存储,以促进我们持续改进和更新。

平台目前支持的生物信息分析流程共7个:
流程名称
功能描述
16S rRNA-QIIME2-Deblur-基础流程
基于QIIME2和Deblur算法搭建的用于16S rRNA基因测序数据分析流程
16S rRNA-QIIME2-DADA2-基础流程
基于QIIME2和DADA2算法搭建的用于16S rRNA基因测序数据分析流程
ITS-QIIME2-Deblur-基础流程
基于QIIME2和Deblur算法搭建的用于ITS测序数据分析流程
ITS-QIIME2-DADA2-基础流程
基于QIIME2和DADA2算法搭建的用于ITS测序数据分析流程
18S rRNA-QIIME2-Deblur-基础流程
基于QIIME2和Deblur算法搭建的用于18S rRNA基因测序数据分析流程
18S rRNA-QIIME2-DADA2-基础流程
基于QIIME2和DADA2算法搭建的用于18S rRNA基因测序数据分析流程
功能基因-QIIME2-DADA2-基础流程
基于QIIME2和DADA2算法搭建的用于功能基因测序数据分析流程,目前已经测试pmoA、nifH和phoD基因。
平台目前支持的生物信息分析模块共5个:
分类
名称
变化趋势分析
小提琴图
箱线图
平均值柱状图(单柱子)
平均值柱状图(多柱子)
平均值散点图(单点)
平均值散点图(多点)
平均值折线图(单线)
平均值折线图(多线)
组间差异检验
ANOSIM检验
MRPP检验
Adonis检验
随机置换检验
Wilcoxon秩和检验
基因/物种组成分析
限制性排序分析(包括RDA、CCA、CAP和dbRDA分析)
非限制性排序分析(包括PCA、PCoA、NMDS和DCA分析)
相对丰度柱状图(分组)
相对丰度柱状图(分面)
相对丰度热图(pheatmap)
相对丰度热图(ggplot2)
相对丰度桑吉图(分组)
相对丰度桑吉图(分面)
相对丰度箱线图
相对丰度饼图
Veen图
相关/回归分析
相关分析(包括Pearson、Spearman和Kendall相关)
Mantel检验(包括mantel检验和偏mantel检验)
向量级回归分析(包括lm和gam回归)
矩阵级回归分析
差异丰度分析
LEfse分析
随机森林
友情链接 Links
关于本站 About us
- 电 话:028-82890506 传真:028-82890288
- Email:lixz@cib.ac.cn 邮 编:610041
- 地 址:中国四川省成都市人民南路四段九号